The Hesperidin Paradox
When a citrus flavonoid binds tighter than synthetic inhibitors—but can't cross a membrane

Type 2 Diabetes Mellitus affects over 500 million people worldwide.1 At its core lies a deceptively simple problem: cells stop listening to insulin. But the molecular machinery governing this communication is anything but simple—it involves a precisely choreographed dance of protein conformations, phosphorylation cascades, and signal transduction pathways. Understanding this machinery at atomic resolution is the first step toward designing better therapeutics.
This research presents a computational investigation of the Human Insulin Receptor (INSR) Kinase Domain, combining structural alignment, sequence conservation analysis, and molecular docking to identify natural compounds that could modulate insulin signaling. The findings suggest that certain plant-derived molecules bind the receptor more tightly than existing synthetic inhibitors.
The Problem: When Cells Stop Responding
Insulin resistance is the metabolic failure mode underlying Type 2 Diabetes.2 When insulin binds its receptor on cell surfaces, it should trigger a cascade that ultimately moves glucose transporters to the membrane, allowing glucose uptake. In insulin-resistant states, this signal gets lost somewhere along the way.
The Human Insulin Receptor is a transmembrane glycoprotein belonging to the receptor tyrosine kinase (RTK) family.3 Structurally, it's a heterotetramer: two extracellular alpha-subunits that bind insulin, connected to two transmembrane beta-subunits containing the intracellular kinase domain. When insulin docks to the alpha-subunits, conformational changes propagate through the membrane, activating the kinase domain through autophosphorylation.4
This kinase domain is the molecular switch. If it fails to change shape correctly, or can't bind its downstream substrates (IRS-1, IRS-2), the entire signaling pathway collapses. No signal means no GLUT4 translocation, no glucose uptake, and chronic hyperglycemia.
The Signaling Cascade
The pathway proceeds through several well-characterized steps:5
- Insulin binding triggers receptor autophosphorylation
- IRS recruitment: Phosphorylated receptor recruits Insulin Receptor Substrate proteins
- PI3K activation: IRS proteins activate Phosphatidylinositol-3-OH kinase
- Akt signaling: PI3K generates PIP3, which activates Protein Kinase B (Akt)
- Downstream effects: Akt phosphorylates targets controlling glucose uptake, glycogen synthesis, and gene expression
In skeletal muscle, this cascade drives GLUT4 translocation through AS160 inhibition and RAC1-mediated actin reorganization.6 In the liver, insulin suppresses gluconeogenesis by phosphorylating FOXO1 (keeping it out of the nucleus) while activating lipogenic programs through SREBP-1c.
Why Computational Approaches?
Traditional experimental methods for studying protein structures—X-ray crystallography, NMR spectroscopy, cryo-EM—are expensive and time-consuming.7 More importantly, they provide static snapshots. Computational methods let us compare dozens of structures simultaneously, quantify subtle conformational differences, and screen thousands of potential drug candidates in silico before committing to wet-lab validation.
The Protein Data Bank (PDB) now contains multiple high-resolution structures of the insulin receptor kinase domain,8 captured in various conformational states: apo (unbound), inhibitor-bound, and phosphorylated. By aligning these structures and measuring their deviations, we can identify which conformations are most suitable for drug design.
Methodology
Structure Selection and Retrieval
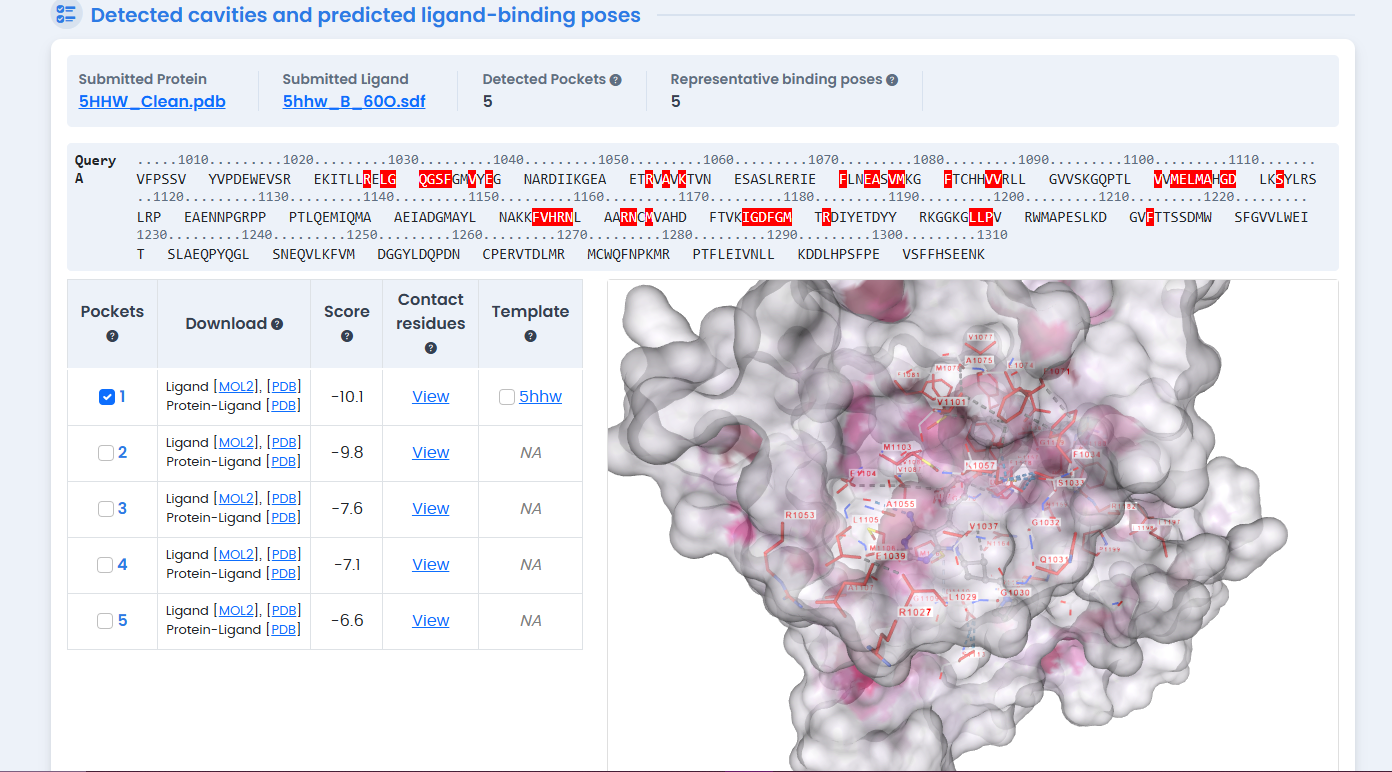
Figure 1: Crystal structure of the Human Insulin Receptor Kinase Domain (PDB ID: 5HHW) with native ligand 60O bound in the ATP-binding pocket. This ligand-bound conformation was selected as the primary template for molecular docking studies.
Seven X-ray crystallographic structures of the INSR Kinase Domain were retrieved from the RCSB Protein Data Bank, all with resolution better than 3.0 Å:9
| PDB ID | Resolution | Description |
|---|---|---|
| 3BU6 | High | Reference structure |
| 3BU3 | High | Apo conformation |
| 5HHW | High | Ligand-bound |
| 4IBM | High | Kinase domain |
| 1IR3 | High | Active conformation |
| 1P14 | High | Phosphorylated |
| 5U1M | Moderate | Fragment |
Structure 3BU6 was selected as the reference template based on its resolution and completeness.
Multiple Sequence Alignment
FASTA sequences from all selected PDB entries were aligned using MultAlin with the BLOSUM62 scoring matrix.10 This revealed the degree of evolutionary conservation across the kinase domain—critical for identifying which structural variations represent genuine conformational flexibility versus experimental artifacts.
The alignment showed high conservation in the catalytic core and ATP-binding pocket, consistent with the tyrosine kinase family's functional constraints.11 This sequence homogeneity suggests that observed structural differences reflect conformational dynamics rather than sequence mutations.
Structural Superposition and RMSD Calculation
Three-dimensional structural alignment was performed in PyMOL using the align command.12 Root Mean Square Deviation (RMSD) values quantify how much atomic positions differ between structures after optimal superposition.
RMSD Results:
| PDB ID | RMSD (Å) | Atoms Aligned | Interpretation |
|---|---|---|---|
| 3BU3 | 0.219 | 2117 | Most stable (best template) |
| 1P14 | 0.465 | 1718 | High similarity |
| 4IBM | 0.608 | 1663 | Good similarity |
| 5HHW | 0.755 | 1831 | Ligand-bound conformation |
| 1IR3 | 0.445 | 1718 | Good similarity |
| 5U1M | 2.621 | 80 | Outlier (truncated fragment) |
The RMSD of 0.219 Å for 3BU3 indicates near-perfect structural identity with the reference—atomic positions differ by less than a quarter of an angstrom across over 2,000 atoms.13 This makes 3BU3 the optimal template for apo-state modeling.
Structure 5HHW, with an RMSD of 0.755 Å, shows moderate deviation reflecting the conformational changes induced by ligand binding. This makes it ideal for docking studies—it represents the receptor in a biologically relevant inhibitor-bound state.
The outlier 5U1M (RMSD 2.621 Å, only 80 atoms aligned) is clearly a truncated fragment unsuitable for whole-domain analysis.
Molecular Docking: Screening Natural Compounds
With structural templates validated, we turned to molecular docking to identify potential inhibitors. The CB-Dock2 server combines curvature-based cavity detection (CurPocket) with AutoDock Vina scoring.14
Target Preparation
The 5HHW structure was prepared for docking by:
- Removing crystallographic water molecules
- Extracting the co-crystallized native inhibitor (Ligand 60O)
- Saving the cleaned receptor structure
CB-Dock2 automatically detected five potential binding cavities. The largest pocket (Volume: 2013 ų) corresponds to the ATP-binding site—the primary target for kinase inhibitors.15
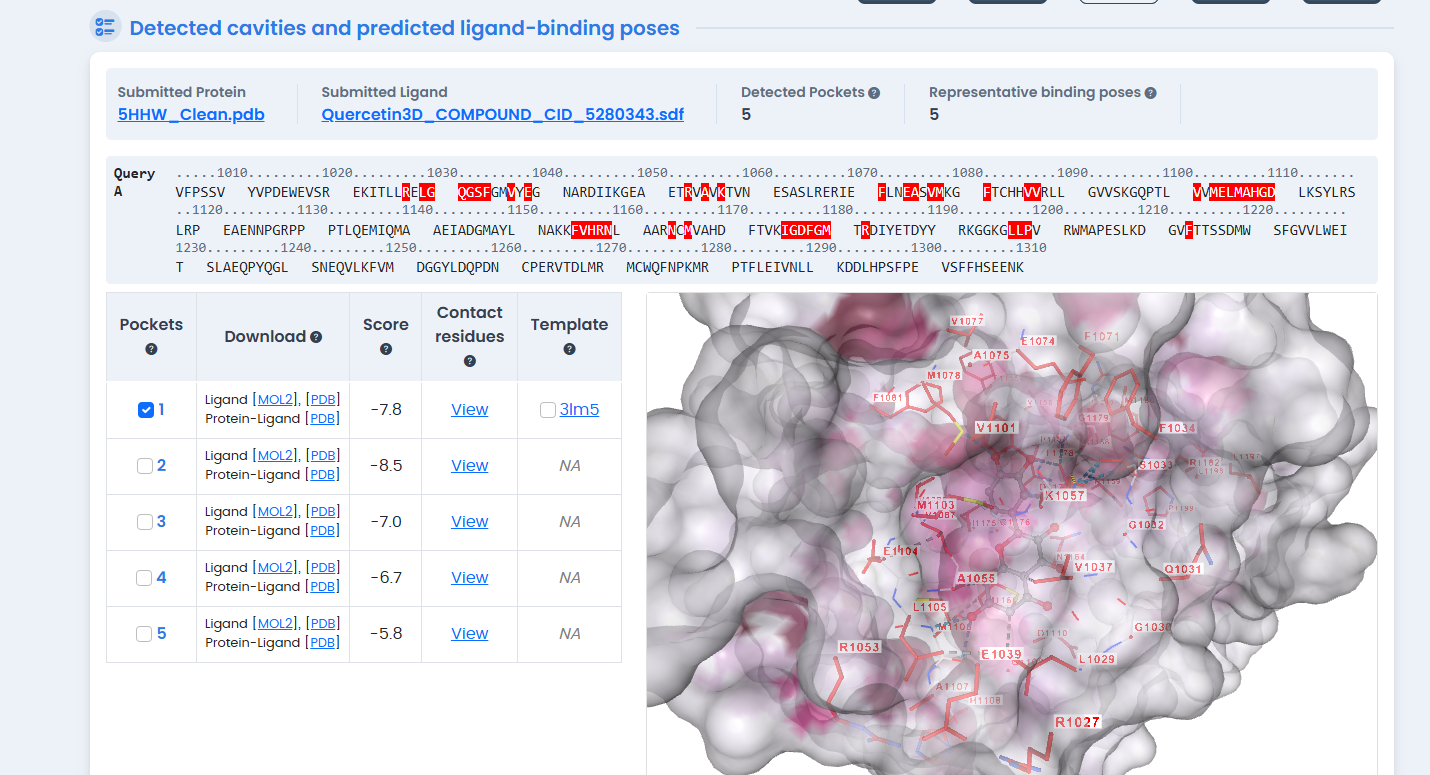
Figure 2: CB-Dock2 blind docking cavity detection on the 5HHW structure. The server automatically identified five potential binding pockets, with the largest cavity (2013 ų) corresponding to the ATP-binding site used for subsequent phytochemical screening.
Docking Validation
Before screening novel compounds, we validated the docking protocol by redocking the native ligand 60O into its original binding site.16 The results:
- Structure-Based Score: -10.1 kcal/mol
- Template-Based Score: -9.9 kcal/mol (Pocket Identity: 1.0)
- Redocking RMSD: 0.0 Å
The perfect RMSD demonstrates that CB-Dock2 can accurately reproduce the experimentally determined binding pose—a critical validation for trusting subsequent predictions.
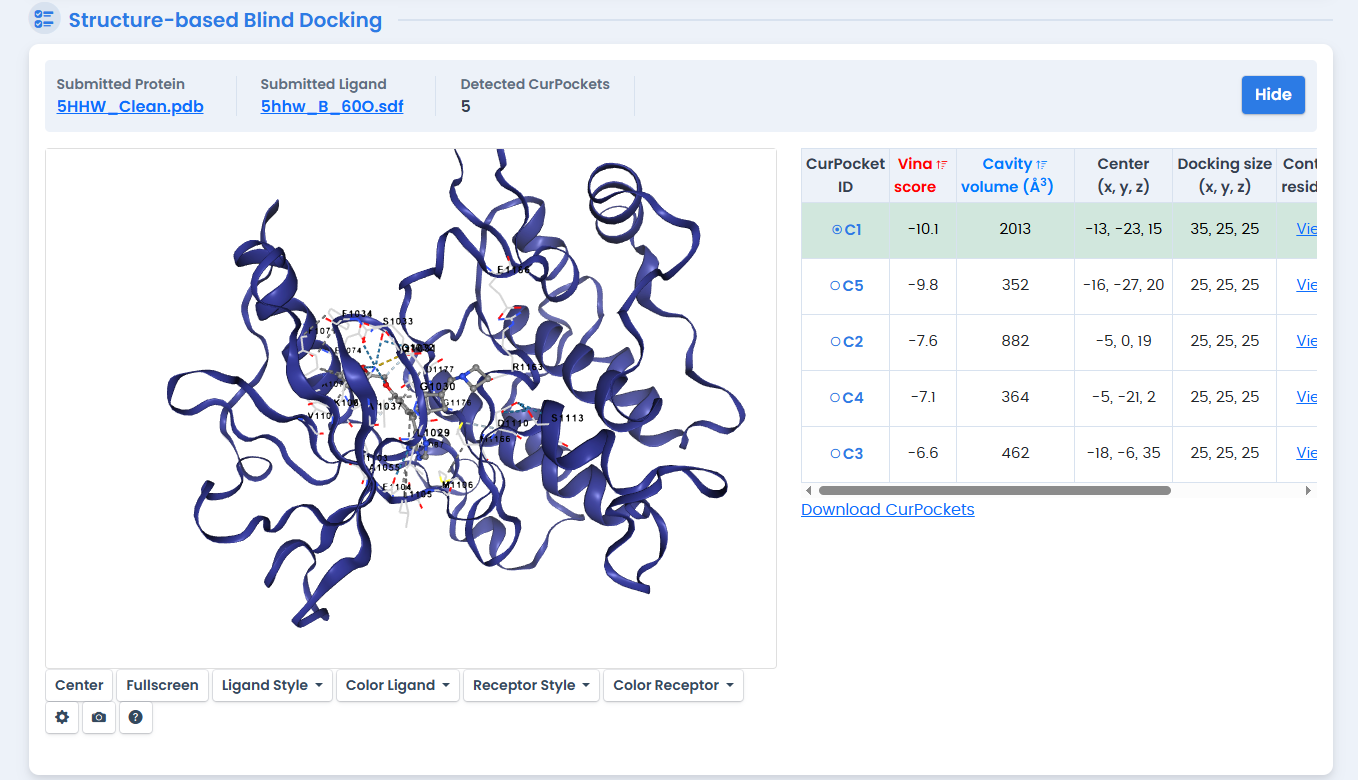
Figure 3: Redocking validation of native ligand 60O using CB-Dock2 structure-based docking. The protocol achieved a Vina score of -10.1 kcal/mol with perfect pose reproduction (RMSD 0.0 Å), confirming the reliability of subsequent docking predictions.
Phytochemical Screening
Five plant-derived compounds with known anti-diabetic or metabolic activity were docked against the validated 5HHW template:17
| Compound | Vina Score (kcal/mol) | Rank | Assessment |
|---|---|---|---|
| Native Ligand 60O | -10.1 | Control | Synthetic inhibitor benchmark |
| Hesperidin | -10.8 | 1st | Outperforms native ligand |
| Quercetin | -8.5 | 2nd | Strong candidate |
| Berberine | -8.4 | 3rd | Strong candidate |
| Curcumin | -7.9 | 4th | Moderate candidate |
| Resveratrol | -7.1 | 5th | Weakest binding |
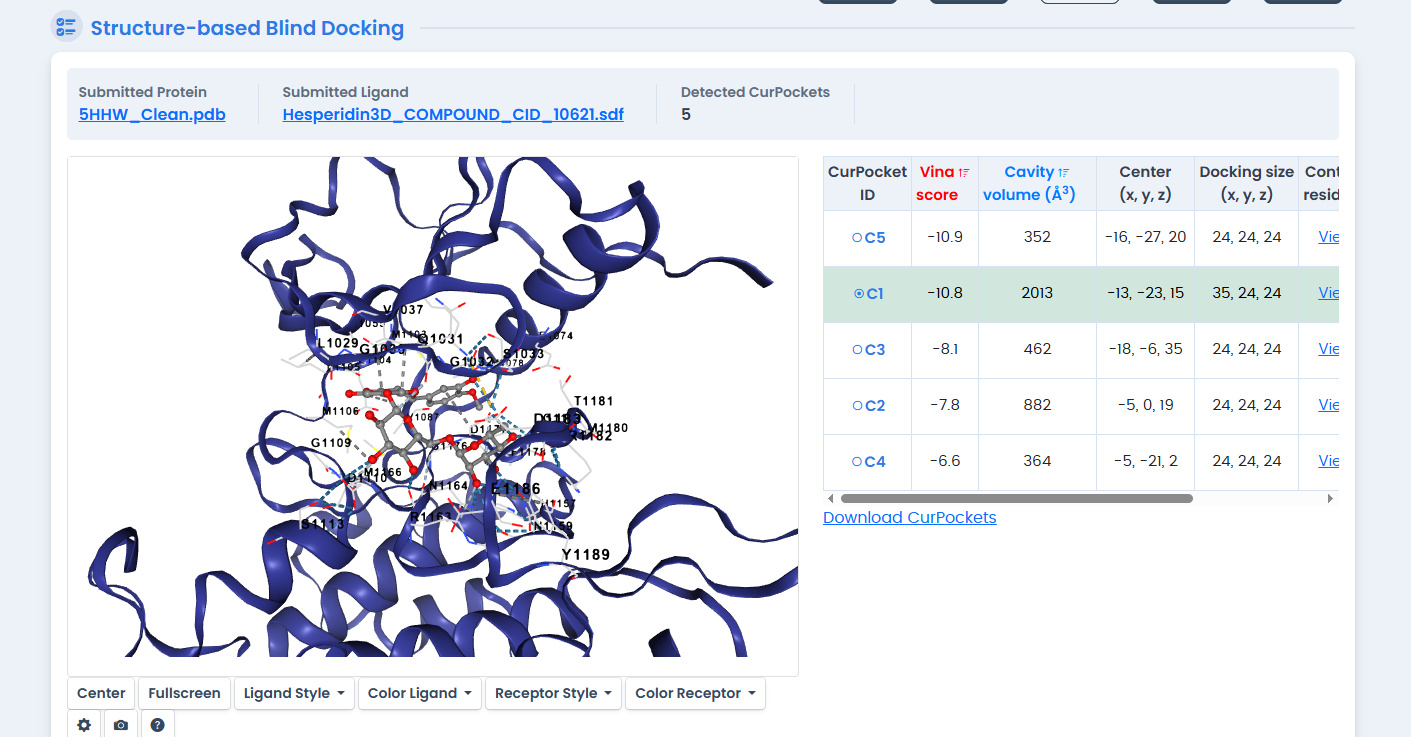
Figure 4: Molecular docking of Hesperidin in the INSR kinase ATP-binding pocket. Hesperidin achieved the highest binding affinity (-10.8 kcal/mol) among all tested compounds, outperforming the synthetic native ligand by 0.7 kcal/mol.
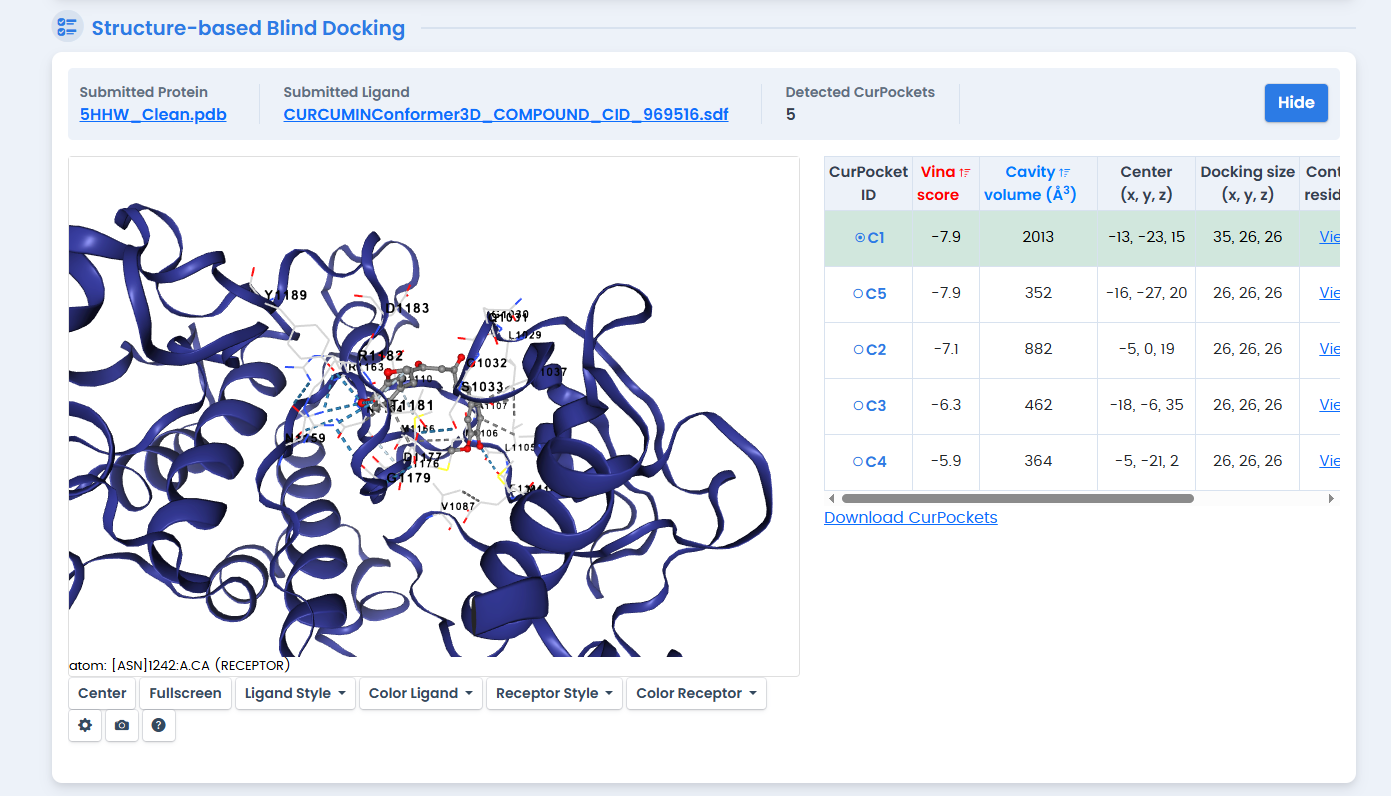
Figure 5: Curcumin docking results showing binding pose within the 5HHW ATP-binding site. Curcumin achieved a Vina score of -7.9 kcal/mol, ranking fourth among the phytochemical candidates.
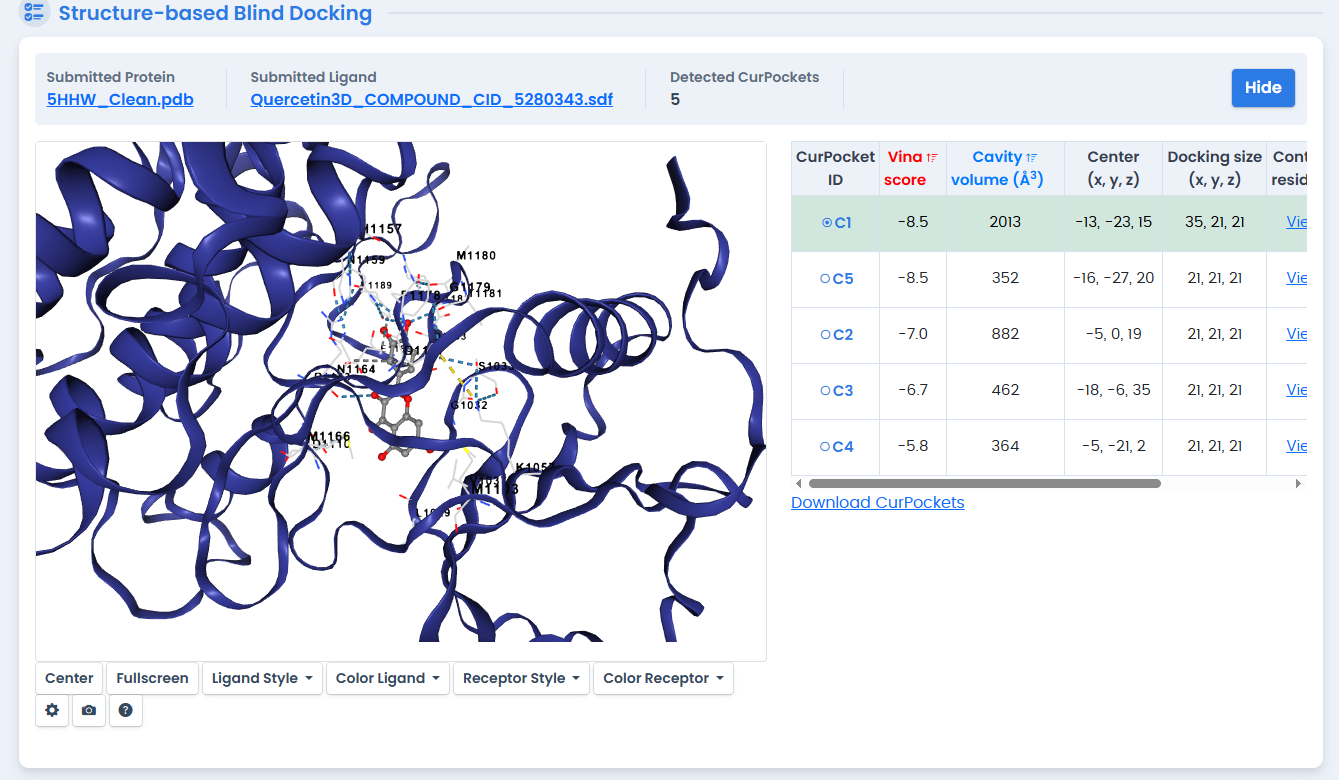
Figure 6: Quercetin molecular docking analysis with the INSR kinase domain. As a flavonoid with established anti-diabetic properties, Quercetin achieved a binding affinity of -8.5 kcal/mol, ranking second among the natural compounds tested.
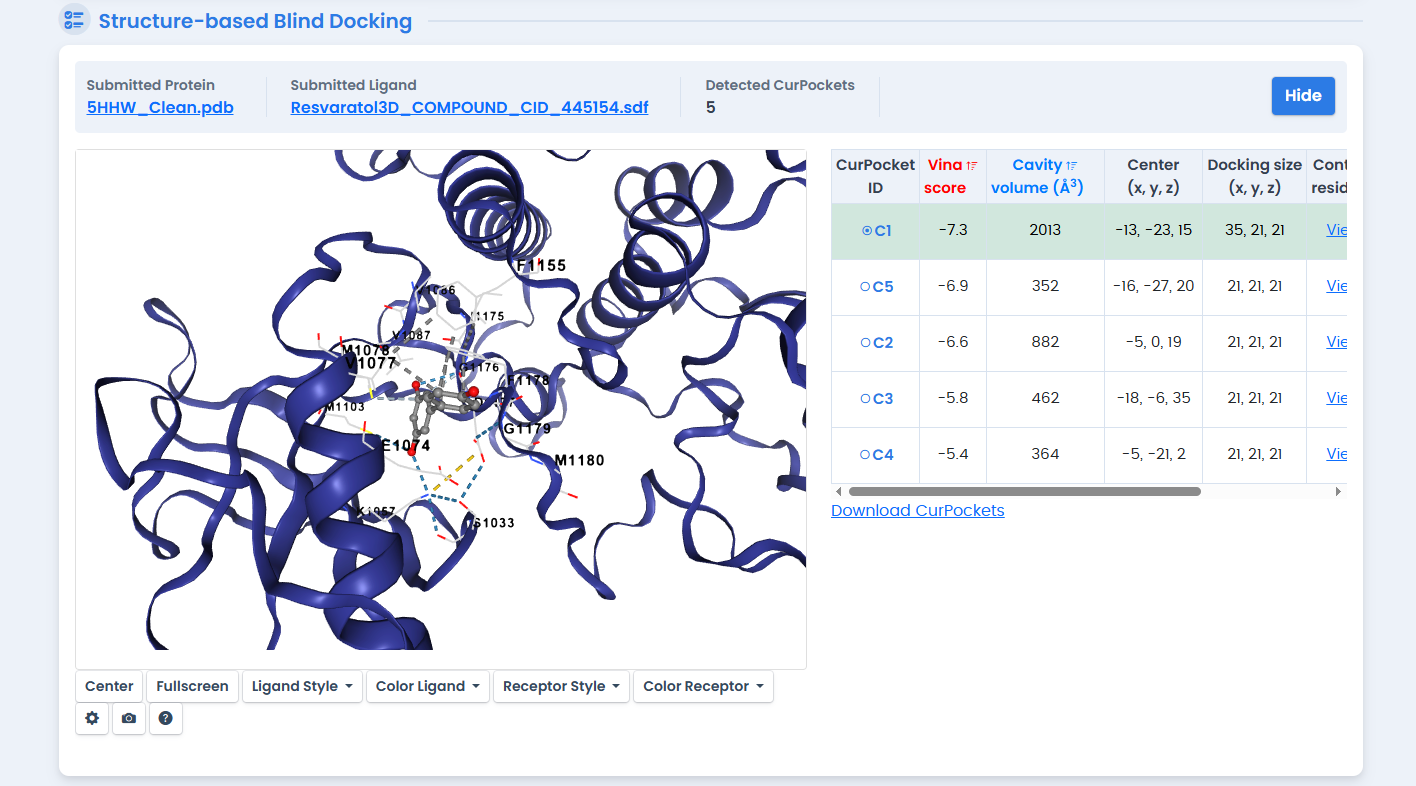
Figure 7: Structure-based docking of Resveratrol against the 5HHW template. Despite its known metabolic benefits, Resveratrol showed the weakest binding affinity (-7.1 kcal/mol) among the five phytochemicals screened.
The standout finding: Hesperidin achieved a binding affinity of -10.8 kcal/mol, surpassing the synthetic inhibitor by 0.7 kcal/mol.18 In binding energy terms, this represents substantially tighter binding—each kcal/mol corresponds to roughly a 5-fold difference in binding constant.
Hesperidin is a flavanone glycoside abundant in citrus fruits.19 Its large molecular structure allows it to form extensive contacts within the ATP-binding pocket, explaining the favorable energetics.
ADMET Profiling: From Binding to Drug-Likeness
Strong binding affinity is necessary but not sufficient for a drug candidate. Compounds must also be absorbed, distributed appropriately, metabolized safely, and excreted efficiently.20 SwissADME analysis revealed important distinctions among the top candidates.
Lipinski's Rule of Five
The classic drug-likeness filter (MW < 500, LogP < 5, H-bond donors < 5, H-bond acceptors < 10):21
| Compound | MW (g/mol) | Violations | Assessment |
|---|---|---|---|
| Berberine | 336.36 | 0 | Fully compliant |
| Curcumin | 368.38 | 0 | Fully compliant |
| Hesperidin | 610.56 | 3 | Multiple violations |
The Hesperidin Paradox
Despite having the highest binding affinity, Hesperidin presents pharmacokinetic challenges:22
- Molecular weight: 610.56 g/mol (exceeds 500 threshold)
- TPSA: 234.29 Ų (far above the 140 Ų limit for oral absorption)
- GI absorption: Predicted low
- BBB permeation: No
The SwissADME Boiled-Egg plot placed Hesperidin outside both the white (GI absorption) and yellow (BBB permeation) regions—flagged as "out of range" for passive diffusion models.
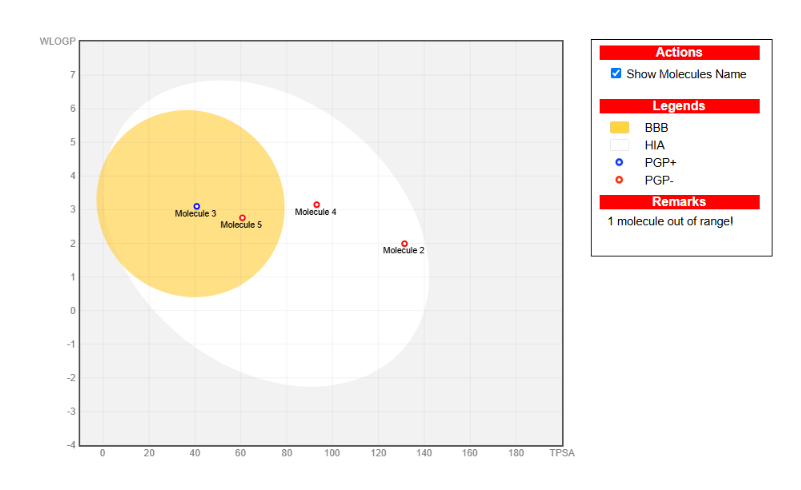
Figure 8: SwissADME Boiled-Egg plot predicting gastrointestinal absorption and blood-brain barrier permeation. Compounds in the white region show high GI absorption; those in the yellow "yolk" can penetrate the BBB. Hesperidin falls outside both regions, indicating poor passive permeability.
This is a common pattern in drug discovery: the most potent binder isn't always the best drug.23 Hesperidin's large glycosidic structure enables tight receptor binding but compromises membrane permeability.
The Berberine Alternative
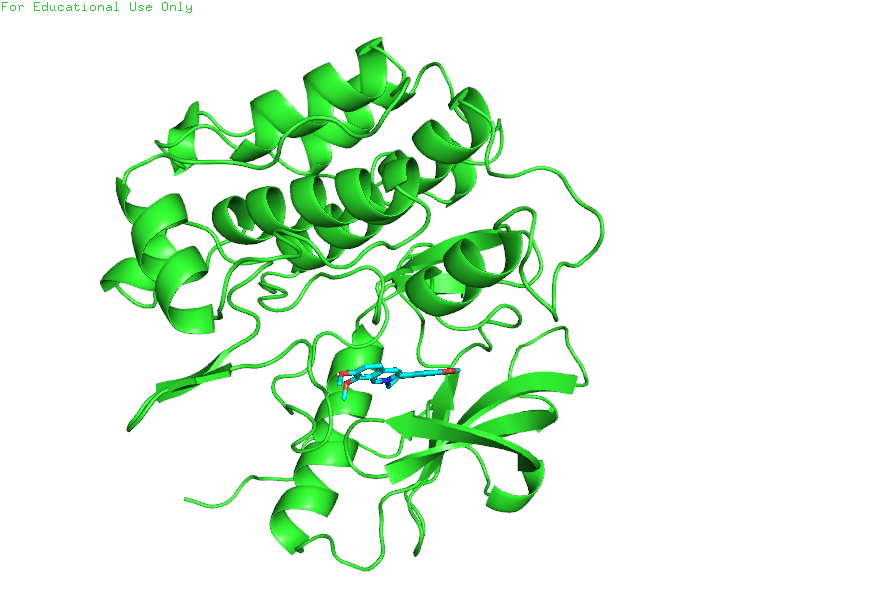
Figure 9: Molecular docking of Berberine in the INSR kinase ATP-binding pocket. Berberine achieved a binding affinity of -8.4 kcal/mol while maintaining full compliance with Lipinski's Rule of Five, making it the most drug-like candidate among the potent binders.
Berberine emerged as the most drug-like candidate:24
- Zero Lipinski violations
- High GI absorption (white region of Boiled-Egg plot)
- BBB permeant (yellow region)
- Bioavailability score: 0.55
- Binding affinity: -8.4 kcal/mol
Its position in the yellow "yolk" indicates both intestinal absorption and blood-brain barrier penetration—relevant for potential CNS effects of insulin signaling modulation.
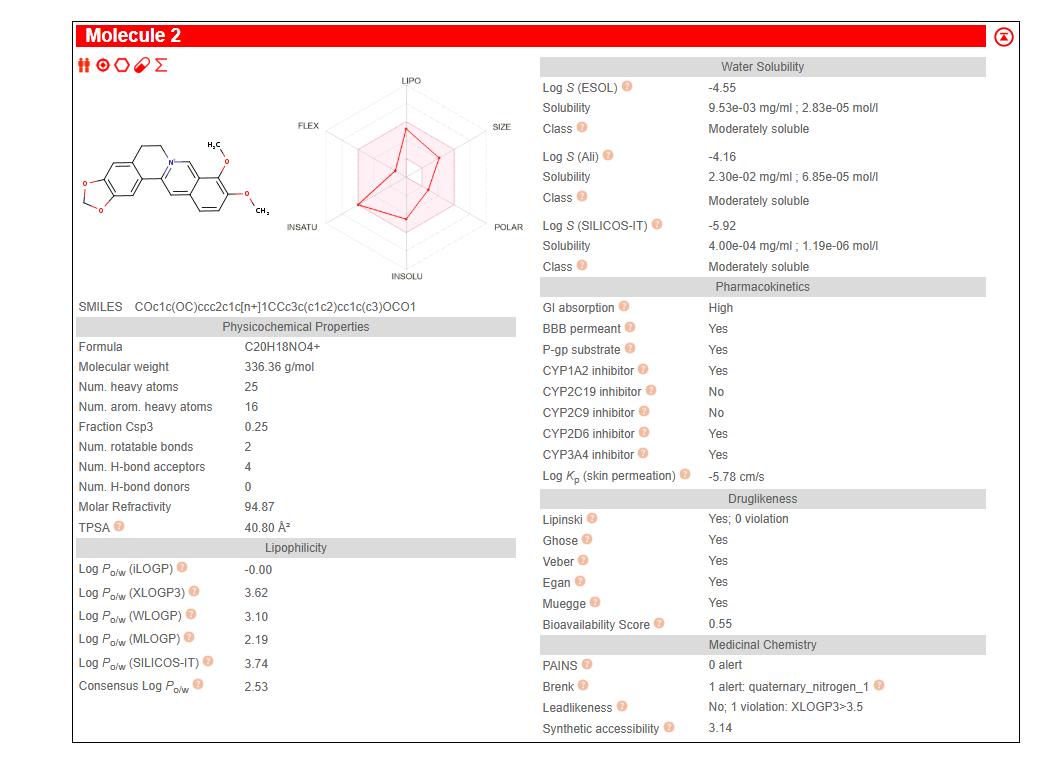
Figure 10: SwissADME pharmacokinetic and drug-likeness profile for Berberine. The compound shows zero Lipinski violations, high gastrointestinal absorption, blood-brain barrier permeability, and a bioavailability score of 0.55—properties that make it an attractive candidate for further development.
Curcumin showed similar drug-likeness (zero violations, high GI absorption) but lacks BBB permeability, making it suitable for peripheral targets only.25
Structural Insights: What the Alignments Reveal
Beyond template selection, the structural analysis revealed functional insights about kinase domain dynamics.
Conservation Patterns
The multiple sequence alignment highlighted near-perfect conservation in:26
- The ATP-binding pocket (glycine-rich loop)
- The catalytic loop containing the critical DFG motif
- The activation loop tyrosine residues
Variations clustered in loop regions distant from the active site—exactly where conformational flexibility is expected and tolerated.
Amino Acid Substitutions
Detailed comparison between structures revealed position-specific substitutions, particularly in the truncated 5U1M fragment. Many substitutions involved polarity changes (polar to non-polar or vice versa), which can affect local folding and solvent accessibility without disrupting the overall kinase fold.27
Ligand-Induced Conformational Changes
The moderate RMSD increase in 5HHW (0.755 Å vs. 0.219 Å for apo 3BU3) reflects genuine structural reorganization upon inhibitor binding.28 The kinase domain doesn't simply accommodate ligands passively—it adapts its conformation to optimize binding interactions.
This induced-fit behavior has implications for drug design: compounds must not only complement the binding pocket's static shape but also be compatible with the pocket's conformational dynamics.
Broader Implications
For Diabetes Therapeutics
Current diabetes drugs like metformin and thiazolidinediones work through various mechanisms but can have significant side effects.29 Direct modulation of insulin receptor signaling represents a potentially more targeted approach.
The identification of Hesperidin as a potent binder suggests that citrus-derived flavonoids warrant further investigation. While Hesperidin itself may require formulation strategies to improve bioavailability (nanoparticles, prodrug approaches, or structural simplification), it provides a starting point for medicinal chemistry optimization.30
Berberine, already used in traditional medicine and available as a supplement, emerges as a particularly attractive candidate given its combination of reasonable binding affinity and favorable drug-likeness.31 Clinical studies of berberine in metabolic disorders have shown promising results, and this structural work provides mechanistic rationale.
For Computational Drug Discovery
This work demonstrates the value of integrating multiple computational approaches:32
- Structural validation (RMSD analysis) ensures reliable templates
- Docking validation (redocking native ligand) confirms protocol accuracy
- ADMET filtering prevents pursuit of non-drug-like compounds
The Hesperidin case illustrates why binding affinity alone is insufficient—pharmacokinetic properties must be considered early in the discovery pipeline.
Limitations and Future Directions
Several caveats apply to these computational predictions:33
Scoring function limitations: Docking scores approximate binding free energies but neglect entropic contributions, protein flexibility, and explicit solvent effects. Experimental validation (ITC, SPR, or enzyme assays) is essential.
Static structures: Crystal structures capture single conformational states. Molecular dynamics simulations could reveal whether identified compounds remain stably bound under physiological conditions.34
Selectivity: The insulin receptor kinase domain shares structural homology with other RTKs. Selectivity profiling against related kinases would be necessary before therapeutic development.
In vivo complexity: Cell-based and animal studies would be needed to confirm that receptor binding translates to functional effects on insulin signaling.
Conclusions
This structural profiling of the Human Insulin Receptor Kinase Domain established:
- Validated templates: PDB ID 3BU3 (RMSD 0.219 Å) for apo-state modeling; 5HHW (RMSD 0.755 Å) for ligand-bound docking
- Protocol validation: Perfect redocking of native ligand (RMSD 0.0 Å, score -10.1 kcal/mol)
- Lead compound: Hesperidin (-10.8 kcal/mol) outperforms the synthetic benchmark
- Drug-like candidates: Berberine and curcumin combine reasonable binding with favorable ADMET profiles
The work provides a foundation for experimental validation and optimization of natural product inhibitors targeting insulin signaling. Whether hesperidin's potency can be maintained while improving its pharmacokinetics—or whether berberine's drug-likeness offers a more practical path forward—remains to be determined through further investigation.
For the hundreds of millions affected by Type 2 Diabetes, every new therapeutic avenue is worth exploring. Computational structural biology accelerates this exploration, filtering thousands of possibilities to identify the few worth pursuing in the laboratory.
This research was conducted at JECRC University, Jaipur, under the supervision of Dr. Dheeraj Chitara, Department of Biotechnology.